
Pharmacogenomics - Precision genetic screen analysis by novel advanced approaches
Our company has made a step forward in the genetic screen analysis, and we have recently proposed well established Molecular Dynamics (MD) methodologies in the investigation of the impact of mutations already classified as disease-causing and those classified as VOUS in the protein structure and function. MD simulations are largely employed in protein structure investigations, but they had been mostly neglected in VOUS classification till now due to their computational expenses and due to the fact that they cannot be performed automatically.
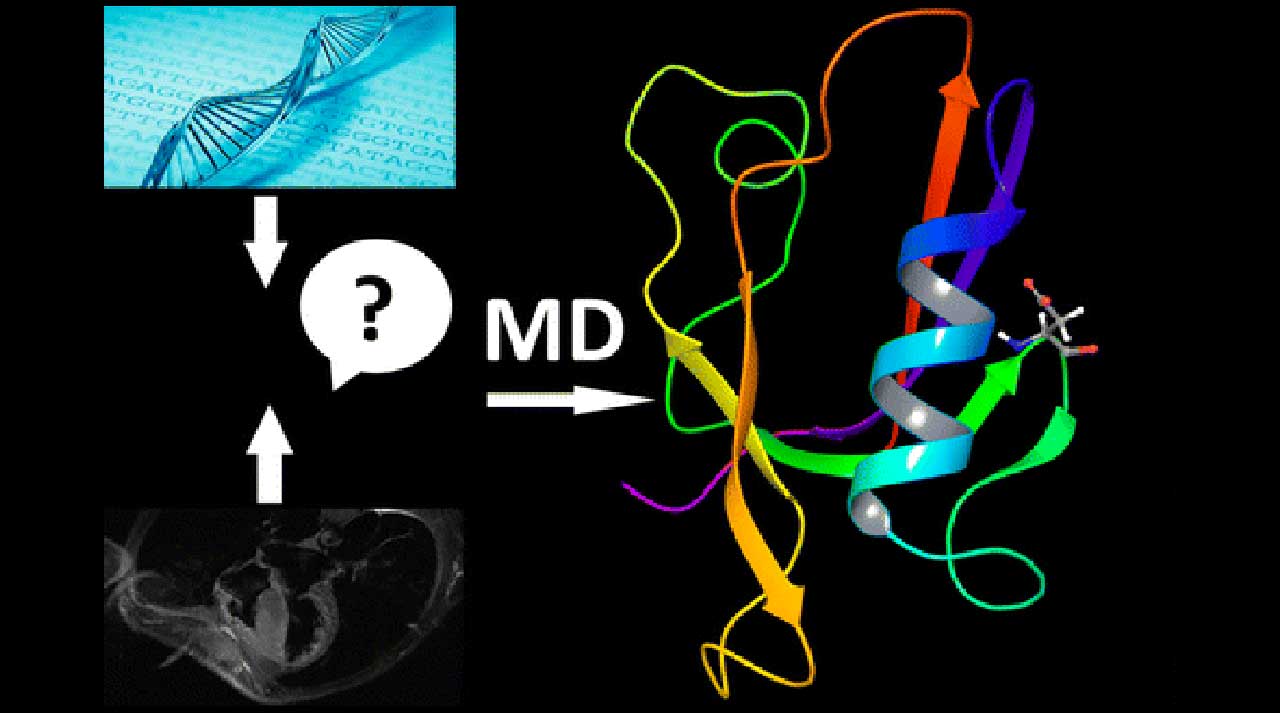
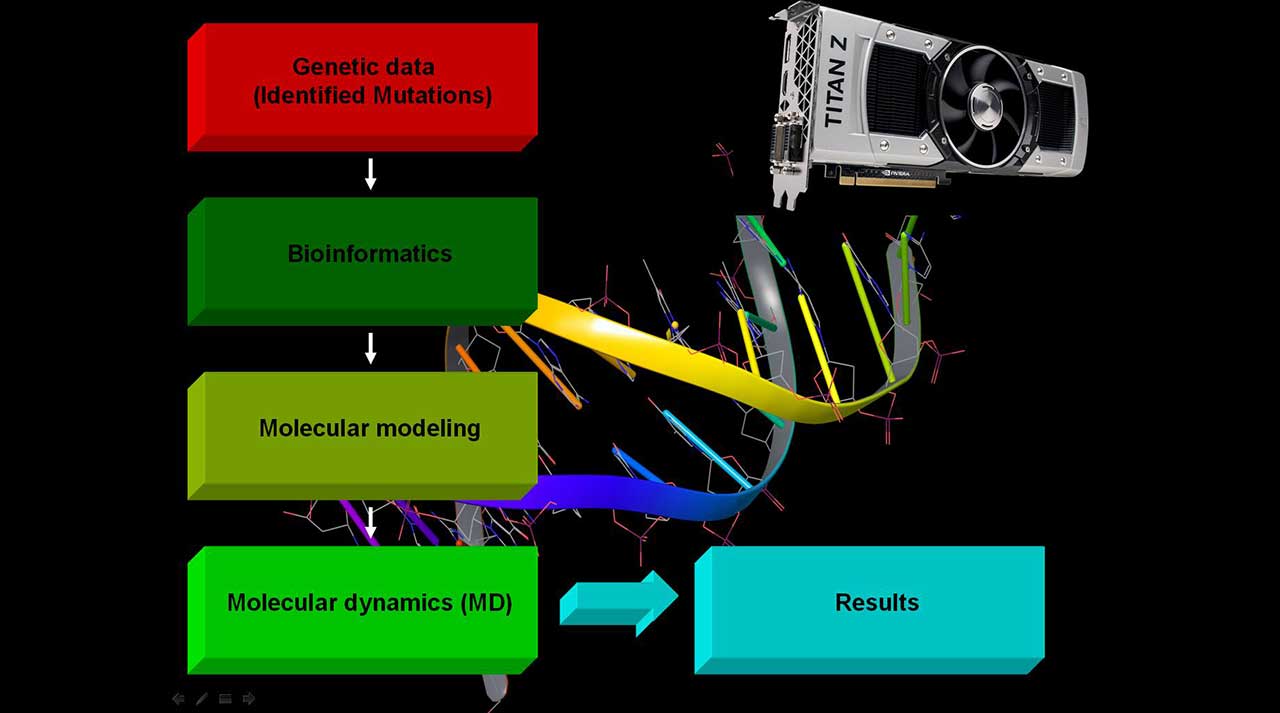
Such a deep analysis brings about the necessity of an individual research project for each particular case, whereas the results have to be interpreted by a highly educated specialist. However, recently, MD studies have become more accessible due to the transfer of calculations on GPU units that increased the speed by a factor of 10 to 30 times and the necessary time to be performed decreased from months to a few days or a week. To put in simple words, MD simulations enable us to reproduce in a realistic way protein behavior in a physiological condition either with an identified mutation or in a native form. Thus, we can answer the question and explain at a molecular level how a mutation changes a certain protein structure and, consequently, its function and, more importantly, its impact on the interactions with other protein partners. This information provides the opportunity for the best treatment, including experimental one, to be chosen by physicians.
We have already tested this approach and used the combination of bioinformatics and MD simulations to reveal the cause of Hypertrophic cardiomyopathy (HCM) of one of our colleagues who has several identified VOUS. A nucleotide change c.160G>A (Gly54Ser) in LDB3 gene (PDZ domain of ZASP protein) was initially identified among 50 genes; we applied an in silico MD and free energy investigations, and demonstrated that G54S on ZASP had a significant structural impact on the formation of α-actinin2−ZASP and LTCC−ZASP complexes [1]. This information, in combination with the adult’s clinical data, indicated that the most likely reason for his HCM was the decreased Ca+ abundance in the heart muscle cells due to the LTCC−ZASP complexes distortion (LTCC is a calcium ion channel). Our results were published in one of the most prestigious journals of the American Chemical Society (ACS) [1], and have already been cited by some high level clinical genetics journals. Importantly, these data were shared with our colleague’s cardiologist, who suggested an experimental therapy of this untreated disease.
Another success of our company in this scientific area was revealing the structural basis of MYH7 (Myosin protein) exons 15–16 HCM mutations and providing directions for drug targeting [2]. Previously, we had also contributed to the knowledge about breast cancer mutations in BRAF protein [3].
Hence, we decided to offer such services to our clients, both from the academia and among the millions of ordinary people who suffer from some genetically linked diseases.
The picture above schematically presents the service which we offer. As an input, we need information about what mutation has been found by your blood genetic test and in which gene. It can be either classified as a disease-causing one or identified as a VOUS. Providing clinical data would also be helpful. Based on these data, we can start the above described analyses of the protein corresponding to a certain gene.
[1] Filip Fratev, Elina Mihaylova, Ilza Pajeva
Combination of genetic screening and molecular dynamics as a useful tool for identification of disease-related mutations: ZASP PDZ domain G54S mutation case;
Journal of Chemical Information and Modeling; 04/2014
[2] Filip Fratev, Svava Ósk Jónsdóttir, Ilza Pajeva;
Structural insight into the UNC-45-Myosin complex.;
Proteins Structure Function and Bioinformatics; 02/2013
[3] Filip Fratev, Svava Ósk Jónsdóttir, Elina Mihaylova and Ilza Pajeva
Molecular Basis of Inactive B-RAFWT and B-RAFV600E Ligand Inhibition, Selectivity and Conformational Stability: An in Silico Study
Mol. Pharmaceutics, 2009, 6 (1), pp 144–157