Structure based in silico drug design - screening for biological active compounds
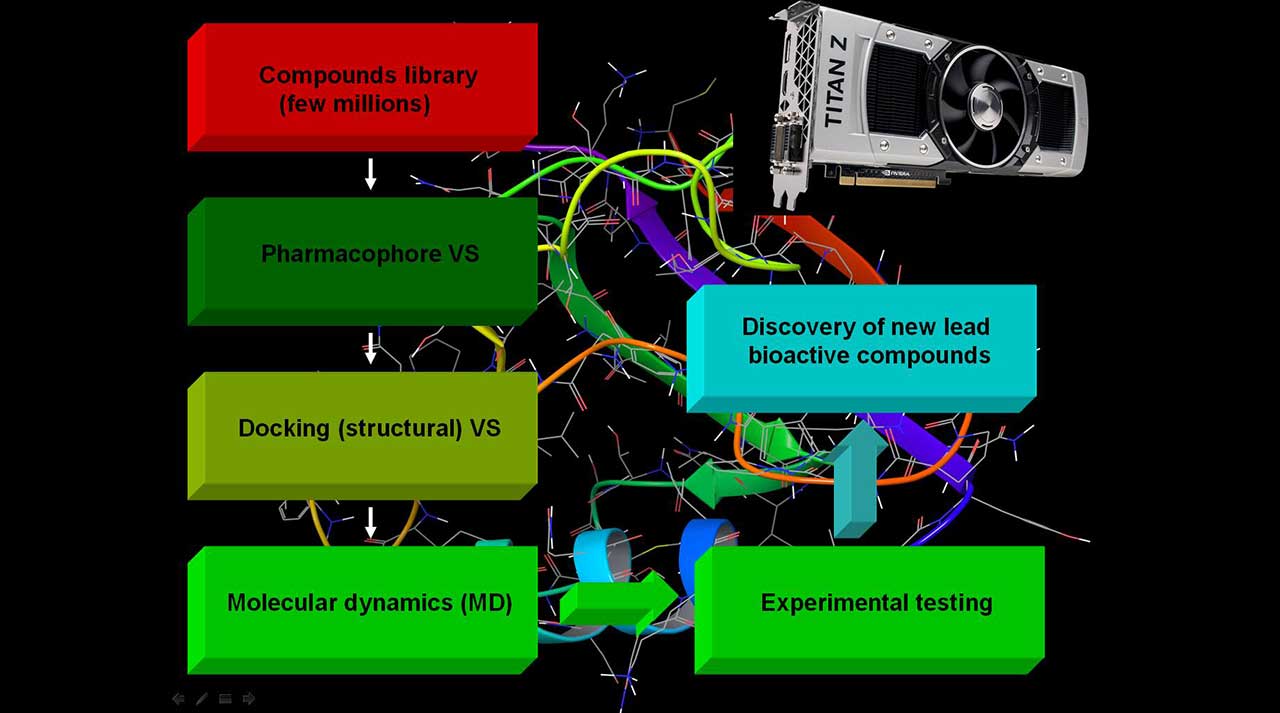
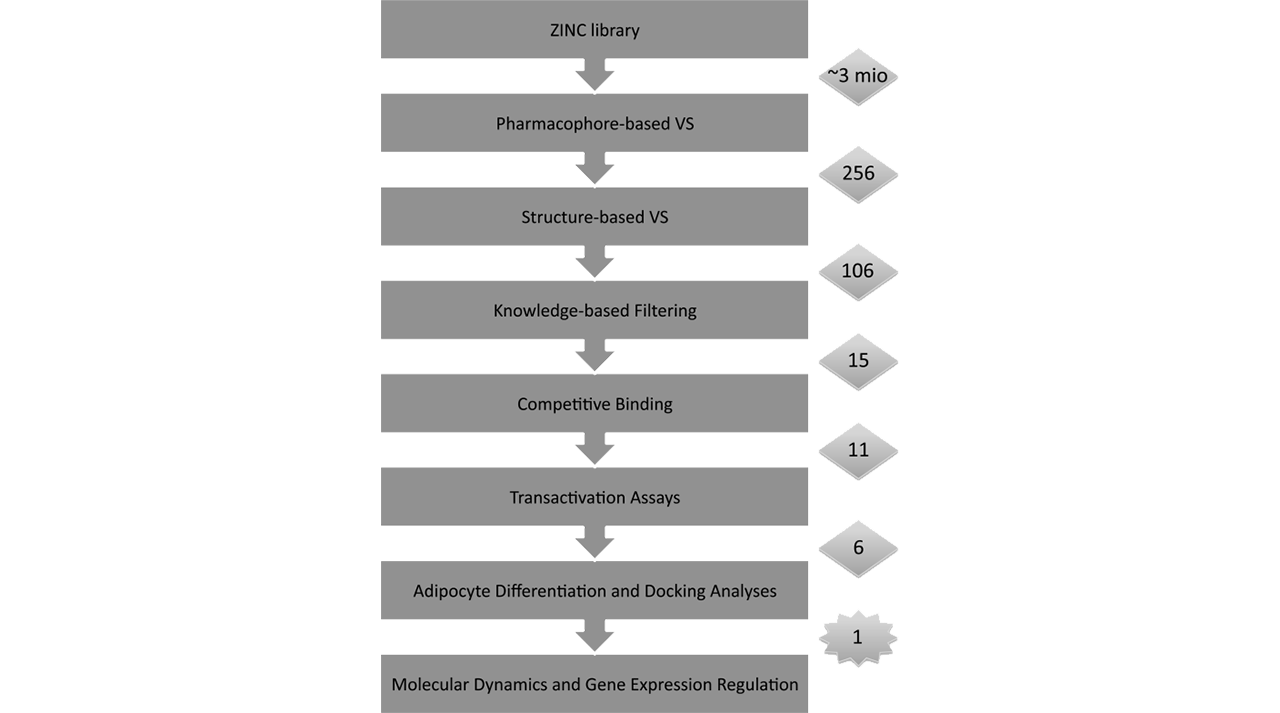
For the structure based in silico screening discovery of biological active compounds, we offer a combination of the most advanced approaches currently employed for such kind of projects. For the last few years it has become more evident that the combination of pharmacophore (ligand based) and docking screening (structure based) approaches is more successful than the use of only one of them. Thus, as an initial step in the screening of millions of compounds (for example ZINC database), as shown in the figure on the right, we combine the techniques above to reduce the potential active compounds to a few hundreds only. Docking is a method which predicts the preferred orientation of a molecule to a protein, when bound to each other to form a stable complex, as well as the strength of the association between them. Pharmacophore models explains how structurally diverse ligands can bind to a common receptor site. Both Docking and Pharmacophore are commonly used in screening novel bioactive ligands. Further, we re-dock the compounds in a more accurate way to the target proteins and then apply diverse advanced Molecular Dynamics (MD) methods to find the most promising bioactive chemicals. Finally, we send the selected compounds for experimental testing and, if requested, lead optimization. It is not a secret that the lead optimization is the most expensive step in any drug design project. Thus, the prediction of the ligands affinity in the frame of about 1 kcal/mol is an important factor. To meat these standards our team provide state of art approaches based on Free Energy of Perturbation (FEP) method. The FEP method has been greatly improved for the last year and can be applied now to most of the industrial projects. It is also well known that the FEP is the most precise method that gather an information for both the ligand selectivity and in the case of drug resistance. The combination of the much improved force field and the speed up of the computations on GPU's are the core of the recent achievements of this precise method.
By this approach we have been able, together with our partners, to discover new classes of partial PPARγ agonists [1].
[1] Irene Kouskoumvekaki, Rasmus K Petersen, Filip Fratev et. all.;
Discovery of a novel selective PPARγ ligand with partial agonist binding properties by integrated in silico/in vitro work flow.
Journal of Chemical Information and Modeling 02/2013